Основна реакція заміщення у водних розчинах - обмін молекул води (22) - була вивчена для великої кількостііонів металів (рис. 34). Обмін молекул води координаційної сфери іона металу з основною масою молекул води, що є розчинником, для більшості металів протікає дуже швидко, і тому швидкість такої реакції вдалося вивчити головним чином методом релаксації. Метод полягає у порушенні рівноваги системи, наприклад, різким підвищенням температури. За нових умов (вищій температурі) система вже не перебуватиме в рівновазі. Потім вимірюють швидкість встановлення рівноваги. Якщо можна змінити температуру розчину протягом 10 -8 сек, можна виміряти швидкість реакції, яка вимагає для свого завершення проміжку часу більше ніж 10 -8 сек.
Можна також виміряти швидкість заміщення координованих молекул води у різних іонів металів лігандами SO 2- 4 , S 2 O 3 2- , EDTA та ін. (26). Швидкість такої реакції
залежить від концентрації гідратованого іону металу і не залежить від концентрації вхідного ліганду, що дозволяє використовувати для опису швидкості цих систем рівняння першого порядку (27). У багатьох випадках швидкість реакції (27) для даного іону металу не залежить від природи вхідного ліганду (L), чи це молекули H 2 O або іони SO 4 2- , S 2 O 3 2- або EDTA.
Це спостереження, а також той факт, що рівняння швидкості цього процесу не включена концентрація вхідного ліганду, дозволяють припускати, що ці реакції протікають по механізму, в якому повільна стадія полягає в розриві зв'язку між іоном металу і водою. З'єднання, що виходить, ймовірно, потім швидко координує знаходяться поблизу ліганди.
У розд. 4 цієї глави було зазначено, що більш високозаряджені гідратовані іони металу, такі, як Al 3+ і Sc 3+ обмінюють молекули води повільніше, ніж іони M 2+ і M + ; це дає підставу припускати, що у стадії, що визначає швидкість всього процесу, важливу роль грає розрив зв'язків. Висновки, отримані в цих дослідженнях, не є остаточними, але вони дають підставу думати, що в реакціях заміщення гідратованих іонів металів важливе значення мають S N 1-процеси.
Ймовірно, найбільш вивченими комплексними сполуками є аміни кобальту (III). Їхня стійкість, легкість приготування та повільно поточні з ними реакції роблять їх особливо зручними для кінетичного вивчення. Оскільки дослідження цих комплексів було проведено лише у водних розчинах, спочатку слід розглянути реакції цих комплексів з молекулами розчинника - води. Було встановлено, що молекули аміаку або амінів, координовані іоном Co(III), настільки повільно заміщуються молекулами води, що зазвичай розглядають заміщення інших лігандів, а не амінів.
Було вивчено швидкість реакцій типу (28) і знайдено, що вона першого порядку щодо комплексу кобальту (X - один із безлічі можливих аніонів).
Оскільки у водних розчинах концентрація H 2 O завжди дорівнює приблизно 55,5 М, не можна визначити вплив зміни концентрації молекул води на швидкість реакції. Рівняння швидкості (29) та (30) для водного розчинуекспериментально не помітні, тому що до просто одно k" = k". Отже, за рівнянням швидкості реакції не можна сказати, чи H 2 O братиме участь у стадії, що визначає швидкість процесу. Відповідь питанням, чи протікає ця реакція механізму S N 2 із заміною іона X на молекулу H 2 O чи механізму S N 1, що передбачає спочатку дисоціацію з наступним приєднанням молекули H 2 O, необхідно отримати з допомогою інших експериментальних даних.
Розв'язання цього завдання можна досягти двома типами експериментів. Швидкість гідролізу (заміщення одного іона Cl – на молекулу води) транс- + приблизно 10 3 разів більше швидкості гідролізу 2+ . Збільшення заряду комплексу призводить до посилення зв'язків метал – ліганд, а отже, і до гальмування розриву цих зв'язків. Слід також враховувати тяжіння вхідних лігандів та полегшення перебігу реакції заміщення. Оскільки виявлено зменшення швидкості зі збільшенням заряду комплексу, то цьому випадку здається найімовірнішим дисоціативний процес (S N 1).
Інший спосіб доказу заснований на вивченні гідролізу серії подібних комплексів транс- +. У цих комплексах молекула етилендіаміну замінена аналогічними діамінами, в яких атоми водню атома вуглецю заміщені на групи CH 3 . Комплекси, що містять заміщені діаміни, реагують швидше, ніж етилендіамінний комплекс. Заміна атомів водню на CH 3 -групи збільшує обсяг ліганду, що ускладнює атаку атома металу іншим лігандом. Ці стеричні перешкоди уповільнюють реакцію механізму S N 2. Наявність поблизу атома металу об'ємистих лігандів сприяє дисоціативному процесу, оскільки видалення однієї з лігандів знижує їх скупчення в атома металу. Спостережуване збільшення швидкості гідролізу комплексів з об'ємними лігандами є хорошим доказом протікання реакції механізму S N 1.
Отже, внаслідок численних досліджень ацидоамінних комплексів Co(II) виявилося, що заміна ацидогруп молекулами води є за своїм характером дисоціативним процесом. Зв'язок атом кобальту - ліганд подовжується до деякої критичної величини, перш ніж молекули води почнуть входити в комплекс. У комплексах, що мають заряд 2+ і вище, розрив зв'язку кобальт - ліганд дуже утруднений, і входження молекул води починає відігравати важливішу роль.
Було виявлено, що заміна ацидо-групи (Х -) в комплексі кобальту(III) іншу групу, ніж молекула H 2 O, (31) проходить спочатку через заміщення її молекулою
розчинника - води з наступною заміною на нову групу Y (32).
Таким чином, у багатьох реакціях з комплексами кобальту(III) швидкість реакції (31) дорівнює швидкості гідролізу (28). Лише іон гідроксилу відрізняється від інших реагентів щодо реакційної здатності з аммінами Co(III). Він дуже швидко реагує з аммінними комплексами кобальту(III) (приблизно в 10 6 разів швидше, ніж вода) за типом реакції основного гідролізу (33).
Знайдено, що ця реакція першого порядку щодо заміщуючого ліганду OH - (34). Загальний другий порядок реакції і незвичайно швидке протікання реакції дозволяють припустити, що іон OH - виключно ефективний нуклеофільний реагент по відношенню до комплексів Co(III) і що протікає реакція за механізмом S N 2 через утворення проміжного з'єднання.
Однак це властивість OH - можна пояснити і іншим механізмом [рівняння (35), (36)]. В реакції (35) комплекс 2+ веде себе як кислота (за Бренстедом), даючи комплекс + , який є амідо-(містить )-сполукою - основою, що відповідає кислоті 2+ .
Потім реакція протікає механізмом S N 1 (36) з утворенням п'ятикоординаційного проміжного з'єднання, далі реагує з молекулами розчинника, що призводить до кінцевого продукту реакції (37). Цей механізм реакції узгоджується зі швидкістю реакції другого порядку і відповідає механізму S N 1. Оскільки реакція у стадії, що визначає швидкість, включає основу, пов'язану з початковим комплексом - кислотою, то цьому механізму дано позначення S N 1СВ.
Визначити, який із цих механізмів найкраще пояснює експериментальні спостереження, дуже важко. Проте є переконливі докази, що підтверджують гіпотезу N 1CB. Найкращі аргументи на користь цього механізму такі: октаедричні комплекси Со(III) взагалі реагують за дисоціативним механізмом S N 1, і немає жодних переконливих доводів, чому б іон OH - повинен зумовити процес S N 2. Встановлено, що іон гідроксилу - слабкий нуклеофільний реагент у реакціях з Pt(II), і тому здається безпричинною його незвичайна реакційна здатність до Co(III). Реакції зі сполуками кобальту(III) у неводних середовищах є прекрасним доказом утворення п'ятикоординаційних проміжних сполук, що передбачаються механізмом S N 1 СВ.
Остаточним доказом є те що, що за відсутності у комплексі Co(III) зв'язків N - Н він повільно реагує з іонами ОН - . Це, звичайно, дає підставу вважати, що для швидкості реакції кислотно-основні властивості комплексу важливіші за нуклеофільні властивості ВІН". Ця реакція основного гідролізу аммінних комплексів Со(III) є ілюстрацією того факту, що кінетичні дані часто можна інтерпретувати не тільки одним способом, Щоб виключити той чи інший можливий механізм, потрібно здійснити досить тонкий експеримент.
В даний час досліджено реакції заміщення великої кількості октаедричних сполук. Якщо розглянути їх механізми реакцій, найчастіше зустрічається дисоціативний процес. Цей результат є несподіваним, оскільки шість лігандів залишають мало місця навколо центрального атома для приєднання до нього інших груп. Відомо лише кілька прикладів, коли доведено виникнення семикоординаційного проміжного з'єднання або виявлено вплив ліганду, що впроваджується. Тому S N 2 механізм не можна повністю відкинути як можливий шлях реакцій заміщення в октаедричних комплексах.
Одна з найважливіших стадій у металокомплексному каталізі – взаємодія субстрату Y з комплексом – відбувається за трьома механізмами:
а) Заміщення ліганду розчинником. Зазвичай таку стадію зображають як дисоціацію комплексу
Суть процесу в більшості випадків - заміщення ліганду L розчинником S, який далі легко замінюється молекулою субстрату
б) Приєднання нового ліганду за вільною координатою з утворенням асоціату з подальшою дисоціацією заміщуваного ліганду
в) Синхронне заміщення (типу S N 2) без утворення інтермедіату
У разі комплексів Pt(II) дуже часто швидкість реакції описується двомаршрутним рівнянням
де k Sі k Y– константи швидкості процесів, що протікають за реакціями (5) (з розчинником) та (6) з лігандомY. Наприклад,


Остання стадія другого маршруту є сумою трьох швидких елементарних стадій – відщеплення Cl – , приєднання Y та відщеплення молекули H 2 O.
У плоских квадратних комплексах перехідних металів спостерігається транс-ефект, сформульований І.І. Черняєвим – вплив LT на швидкість заміщення ліганду, що знаходиться в положенні до ліганду LT. Для комплексів Pt(II) транс-ефект зростає у ряді лігандів:
H 2 O~NH 3 Наявність кінетичного трансефекту та термодинамічного трансвпливу пояснює можливість синтезу інертних ізомерних комплексів Pt(NH 3) 2 Cl 2: Реакції електрофільного заміщення (S E) водню металом у координаційній сфері металу та зворотні ним процеси SH - H 2 O, ROH, RNH 2 , RSH, ArH, RCCН. Навіть молекули H 2 і CH 4 беруть участь у реакціях такого типу Реакції застосування Lз зв'язкуM-X У разі X = R (металоорганічний комплекс) координовані металом молекули також впроваджуються по зв'язку M-R (L-CO, RNC, C2H2, C2H4, N2, CO2, O2 та ін.). Реакції впровадження є результатом внутрішньомолекулярної атаки нуклеофілаX на координовану по- або -типу молекулу. Зворотні реакції – реакції - та -елімінування Реакції окисного приєднання та відновлювального елімінування M 2 (C 2 H 2) M 2 4+ (C 2 H 2) 4– Мабуть, у цих реакціях завжди має місце попередня координація молекули, що приєднується, але це не завжди вдається зафіксувати. Тому наявність вільного місця в координаційній сфері або місця, пов'язаного з розчинником, який легко замінюється субстратом, є важливим фактором, що впливає на реакційну здатність металокомплексів. Наприклад, біс--алільні комплекси Ni є хорошими попередниками каталітично активних частинок, оскільки внаслідок легкого відновлювального елімінування біс-алілу з'являється комплекс з розчинником, т.зв. "Голий" нікель. Роль вільних місць ілюструє такий приклад: Реакції нуклеофільного та електрофільного приєднання до - та -комплексів металів В якості інтермедіатів каталітичних реакцій зустрічаються як класичні металоорганічні сполуки, що мають зв'язки M-C, M=CіMC, так і некласичні сполуки, в яких органічний ліганд координований по 2 , 3 , 4 , 5 і 6 -типу, є елементом електронно-дефіцитних структур – місткові СН 3 та С 6 Н 6 -групи, некласичні карбіди (Rh 6 C(CO) 16 ,C(AuL) 5 + ,C(AuL) 6 2+ та ін.). Серед специфічних механізмів для класичних -металоорганічних сполук відзначимо кілька механізмів. Так, встановлено 5 механізмів електрофільного заміщення атома металу зв'язку M-C. електрофільне заміщення з нуклеофільним сприянням AdEПриєднання-елімінування AdE(C) Приєднання до атома С вsp 2 -гібридизації AdE(M) Приєднання окисне до металу Нуклеофільне заміщення у атома вуглецю в реакціях деметалювання металоорганічних сполук, відбувається як окисно-відновний процес: Можлива участь окислювача у такій стадії Таким окислювачем може бути CuCl 2 , п-бензохинон,NO 3 – та інших. сполуки. Наведемо ще дві характерні для RMX елементарні стадії: гідрогеноліз зв'язку M-C та гомоліз зв'язку M-C Важливим правилом, що стосується всіх реакцій комплексних і металоорганічних сполук і пов'язаних з принципом найменшого руху, є правило 16-18-електронної оболонки Толмена (розділ 2). Реакції заміщення, приєднання чи відщеплення лігандів, у яких змінюється координаційна сфера металу. У широкому розумінні під реакціями заміщення розуміють процеси заміщення одних лігандів у координаційній сфері металу. Диссоціативний (D) механізм. Двостадійний процес у граничному випадку протікає через інтермедіат із меншим координаційним числом: ML6<->+ L; + Y --» ML5Y Асоціативний (А) механізм. Двостадійний процес, що характеризується утворенням інтермедіату з великим координаційним числом: ML6 + Y = ; = ML5Y + L Механізм взаємного обміну (І). З цього механізму протікає більшість реакцій обміну. Процес одностадійний і супроводжується утворенням інтермедіату. У перехідному стані реагент і група, що йде, пов'язані з реакційним центром, входять до його найближчої координаційної сфери, і в процесі реакції відбувається витіснення однієї групи іншої, обмін двох лігандів: ML6 + Y = = ML5Y+L Внутрішній механізм. Цей механізм характеризує заміщення лігандів на молекулярному рівні. 2. Особливості властивостей лантаноїдів (Ln), пов'язані з ефектом лантаноїдного стиснення. Сполуки Ln 3+: оксиди, гідроксиди, солі. Інші ступеня окиснення. Приклади відновлювальних властивостей Sm 2+ , Eu 2+ та окисних властивостей Ce 4+ , Pr 4+ . Монотонне зменшення атомних та іонних радіусів при русі по ряду 4f-елементів отримало назву лантаноїдного стиснення. я. Воно призводить до того, що радіуси атомів наступних за лантаноїдами 5d-перехідних елементів четвертої (гафній) та п'ятої (тантал) груп виявляються практично рівними радіусам їх електронних аналогів з п'ятого періоду: цирконію та ніобію відповідно, а хімія важких 4d- та 5d-металів має багато спільного. Іншим наслідком f-стиснення є близькість іонного радіусу ітрію до радіусів важких f-елементів: диспрозія, гольмія та ербія. Усі РЗЕ утворюють стійкі оксиди у ступені окислення +3. Вони є тугоплавкі кристалічні порошки, що повільно поглинають вуглекислий газ і пари води. Оксиди більшості елементів отримують прожарюванням на повітрі гідроксидів, карбонатів, нітратів, оксалатів при температурі 800-1000 °С. Утворюють оксиди M2O3 та гідроксиди M(OH)3 Тільки гідроксид скандія амфотерен Оксиди та гідроксиди легко розчиняються в кислотах Sc2O3 + 6HNO3 = 2Sc(NO3)3 + 3H2O Y(OH)3 + 3HCl = YCl3 + 3H2O Тільки з'єднання скандію гідролізуються у водному розчині Cl3 ⇔ Cl2 + HCl Відомі всі галогеніди у ступені окислення +3. Усі – тугоплавки. Фториди погано розчиняються у воді. Y(NO3)3 + 3NaF = YF3↓+ 3NaNO3 Комплексні з'єднання. Їхня будова на основі координаційної теорії А. Вернера. Комплексний іон, його заряд. Катіонні, аніонні, нейтральні комплекси. Номенклатура, приклади. Реакція заміщення лігандів. Константа нестійкості комплексного іона, константа стійкості. До нестійкості-це відношення творів концентрації іонів, що розпалися, на нерозпалу кількість. До уст = 1/ До нест (зворотна величина) 43. Конкуренція за ліганд або за комплексоутворювач: ізольоване та поєднане рівноваги заміщення лігандів. Загальна константа суміщеної рівноваги заміщення лігандів. В результаті конкуренції протон руйнує досить міцний комплекс, утворюючи слабко дисоціюючу речовину - воду. Cl + NiS0 4 +4NH 3 ^ S0 4 +AgCl I Це вже приклад конкуренції ліганду за комплексоутворювач, з утворенням більш міцного комплексу (K H + =9,3-1(Г 8 ; К Н [М(Ш 3) 6 ] 2+ = 1,9-Ю -9) та важкорозчинної сполуки AgCl - K s = 1,8 10" 10 Уявлення про будову металоферментів та інших біокомплексних сполук (гемоглобін, цитохроми, кобаламіни). Фізико-хімічні принципи транспортування кисню гемоглобіном Кобаламіни. Вітаміни B 12називають групу кобальтовмісних біологічно активних речовин, званих кобаламінами. До них відносять власне ціанокобаламін, гідроксикобаламін та дві коферментні форми вітаміну B 12: метилкобаламін та 5-дезоксиаденозилкобаламін Іноді у вужчому сенсі вітаміном B 12 називають ціанокобаламін, так як саме в цій формі в організм людини надходить основна кількість вітаміну B 12 , не зважаючи на те, що він не синонім з B 12 , і кілька інших сполук також мають B 12 - вітамінної активністю. Вітамін B12 також називається зовнішнім фактором Касла. B 12 має найскладнішу порівняно з іншими вітамінами хімічну структуру, основою якої є корінове кільце. Коррін багато в чому схожий на порфірин (складна хімічна структура, що входить до складу гему, хлорофілу і цитохромів), але відрізняється від порфірину тим, що два піррольні цикли у складі корину з'єднані між собою безпосередньо, а не метиленовим містком. У центрі корінової структури розташований іон кобальту. Чотири координаційні зв'язки кобальт утворює з атомами азоту. Ще один координаційний зв'язок з'єднує кобальт з диметилбензімідазольним нуклеотидом. Останній, шостий координаційний зв'язок кобальту залишається вільним: саме з цього зв'язку і приєднується ціаногрупа, гідроксильна група, метильний або 5"-дезоксиаденозильний залишок з утворенням чотирьох варіантів вітаміну B 12 , відповідно. Ковалентний зв'язок Приклад приклад ковалентного зв'язку перехідний метал-вуглець. Глава 17. Комплексні з'єднання 17.1. Основні визначення У цьому розділі ви познайомитеся з особливою групою складних речовин, які називаються комплексними(або координаційними) з'єднаннями. В даний час строгого визначення поняття комплексна часткані. Зазвичай використовується таке визначення. Наприклад, гідратований іон міді 2 - комплексна частка, так як вона реально існує в розчинах і деяких кристалогідратах, утворена з іонів Cu 2 і молекул H 2 O, молекули води - реально існуючі молекули, а іони Cu 2 існують в кристалах багатьох сполук міді. Навпаки, іон SO 4 2 не є комплексною частинкою, оскільки, хоч іони O 2 у кристалах зустрічаються, іон S 6 у хімічних системах не існує. Приклади інших комплексних частинок: 2, 3, 2, 2 . Разом з тим до комплексних частинок відносять іони NH 4 і H 3 O хоча іони H в хімічних системах не існують. Іноді комплексними частинками називають складні хімічні частинки, усі або частина зв'язків у яких утворені за донорно-акцепторним механізмом. У більшості комплексних частинок так і є, але, наприклад, в алюмокалієвих галун SO 4 в комплексній частинці 3 зв'язок між атомами Al і O дійсно утворена по донорно-акцепторного механізму, а в комплексній частинці є лише електростатична (іон-дипольна) взаємодія. Підтвердженням цього є існування в залізоамонійних галунах аналогічної за будовою комплексної частинки , в якій між молекулами води та іоном NH 4 можлива тільки іон-дипольна взаємодія. По заряду комплексні частинки може бути катіонами, аніонами, і навіть нейтральними молекулами. Комплексні сполуки, що включають такі частинки, можуть належати до різних класів хімічних речовин (кислот, основ, солей). Приклади: (H 3 O) – кислота, OH – основа, NH 4 Cl та K 3 – солі. Зазвичай комплексоутворювач – атом елемента, що утворює метал, але може бути і атом кисню, азоту, сірки, йоду та інших елементів, які утворюють неметали. Ступінь окислення комплексоутворювача може бути позитивною, негативною або рівною нулю; при утворенні комплексного з'єднання з найпростіших речовин вона змінюється. Лігандами можуть бути частинки, до утворення комплексного з'єднання молекули (H 2 O, CO, NH 3 та ін), аніони (OH , Cl , PO 4 3 та ін), а також катіон водню. Розрізняють унідентатніабо монодентатні ліганди (пов'язані з центральним атомом через один зі своїх атомів, тобто одним -зв'язком), бідентатні(пов'язані з центральним атомом через два своїх атоми, тобто двома зв'язками), трідентатніі т.д. Якщо ліганди унідентатні, то координаційне число дорівнює кількості таких лігандів. КЧ залежить від електронної будови центрального атома, від його ступеня окиснення, розмірів центрального атома та лігандів, умов утворення комплексної сполуки, температури та інших факторів. КЧ може приймати значення від 2 до 12. Найчастіше воно дорівнює шести, дещо рідше – чотирьом. Існують комплексні частинки та з декількома центральними атомами. Використовуються два види структурних формул комплексних частинок: із зазначенням формального заряду центрального атома та лігандів, або із зазначенням формального заряду всієї комплексної частинки. Приклади: Для характеристики форми комплексної частки використовується уявлення про координаційний поліедр (багатогранник). До координаційних поліедр відносять також квадрат (КЧ = 4), трикутник (КЧ = 3) і гантель (КЧ = 2), хоча ці фігури і не є багатогранниками. Приклади координаційних поліедрів і відповідних форм комплексних частинок для найбільш поширених значень КЧ наведено на рис. 1. 17.2. Класифікація комплексних з'єднань Як хімічні речовини комплексні сполуки поділяються на іонні (їх іноді називають іоногенними) та молекулярні ( неіоногенні) з'єднання. Іонні комплексні сполуки містять заряджені комплексні частинки – іони – є кислотами, основами чи солями (див. § 1). Молекулярні комплексні сполуки складаються з незаряджених комплексних частинок (молекул), наприклад: або віднесення їх до якого-небудь основного класу хімічних речовин важко. Комплексні частинки, що входять до складу комплексних сполук, досить різноманітні. Тому для їхньої класифікації використовується кілька класифікаційних ознак: число центральних атомів, тип ліганду, координаційне число та інші. За кількістю центральних атомівкомплексні частинки поділяються на одноядерніі багатоядерні. Центральні атоми багатоядерних комплексних частинок можуть бути пов'язані між собою або безпосередньо або через ліганди. І в тому, і в іншому випадку центральні атоми з лігандами утворюють єдину внутрішню сферу комплексної сполуки: За типом лігандів комплексні частки поділяються на 1) Аквакомплекси, тобто комплексні частинки, в яких як ліганди присутні молекули води. Більш менш стійкі катіонні аквакомплекси m, аніонні аквакомплекси нестійкі. Всі кристалогідрати відносяться до сполук, що містять аквакомплекси, наприклад: Mg(ClO 4) 2 . 6H 2 O насправді (ClO 4) 2; 2) Гідроксокомплекси, тобто комплексні частинки, в яких як ліганди присутні гідроксильні групи, які до входження до складу комплексної частинки були гідроксид-іонами, наприклад: 2 , 3 , . Гідроксокомплекси утворюються з аквакомплексів, що виявляють властивості катіонних кислот: 2 + 4OH = 2 + 4H 2 O 3) Аміакати, тобто комплексні частинки, в яких як ліганди присутні групи NH 3 (до утворення комплексної частинки - молекули аміаку), наприклад: 2 , 3 . Аміакати також можуть бути одержані з аквакомплексів, наприклад: 2 + 4NH 3 = 2 + 4 H 2 O Забарвлення розчину в цьому випадку змінюється з блакитного до ультрамаринового. 4) Ацидокомплекси, тобто комплексні частинки, в яких як ліганди присутні кислотні залишки як безкисневих, так і кисневмісних кислот (до утворення комплексної частки - аніони, наприклад: Cl , Br , I , CN , S 2 , NO 2 , S 2 O 3 2 , CO 3 2 , C 2 O 4 2 тощо). Приклади утворення ацидокомплексів: Hg 2 + 4I = 2 Остання реакція використовується у фотографії для видалення з фотоматеріалів срібла, що не прореагував броміду. 5) Комплекси, в яких лігандами є атоми водню, поділяються на дві різні групи: гідриднікомплекси та комплекси, що входять до складу онієвихз'єднань. При утворенні гідридних комплексів – , , – центральний атом є акцептором електронів, а донором – гідридний іон. Ступінь окиснення атомів водню у цих комплексах дорівнює –1. У онієвих комплексах центральний атом є донором електронів, а акцептором – атом водню у ступеня окиснення +1. Приклади: H 3 O або – іон оксонію, NH 4 або – іон амонію. Крім того, існують і заміщені похідні таких іонів: – іон тетраметиламонію, – іон тетрафеніларсонію, – іон діетілоксонію тощо. 6) Карбонільнікомплекси – комплекси, у яких як лігандів присутні групи CO (до утворення комплексу – молекули монооксиду вуглецю), наприклад: , , та інших. 7) Аніонгалогенатнікомплекси - комплекси типу. За типом лігандів виділяють інші класи комплексних частинок. Крім того, існують комплексні частинки з різними за типом лігандами; Найпростіший приклад - аква-гідроксокомплекс. 17.3. Основи номенклатури комплексних сполук Формула комплексної сполуки складається так само, як і формула будь-якої іонної речовини: на першому місці записується формула катіону, на другому – аніону. Формула комплексної частки записується в квадратних дужках у наступній послідовності: на першому місці ставиться символ елемента-комплексоутворювача, далі – формули лігандів, що були до утворення комплексу катіонами, потім – формули лігандів, що були до утворення комплексу нейтральними молекулами, і після них – формули лігандів, що були до утворення комплексу аніонами. Назва комплексної сполуки будується так само, як і назва будь-якої солі або основи (комплексні кислоти називаються солями водню або оксонію). У назву з'єднання входить назва катіону та назва аніону. У назву комплексної частки входить назва комплексоутворювача та назви лігандів (назва записується відповідно до формули, але справа наліво. Для комплексоутворювачів у катіонах використовуються російські назви елементів, а в аніонах – латинські. Назви найпоширеніших лігандів: Приклади назв комплексних катіонів: Приклади назв комплексних аніонів: 2 – тетрагідроксоцінкат-іон Приклади назв нейтральних комплексних частинок: Більш докладні номенклатурні правила наводяться у довідниках та спеціальних посібниках. 17.4. Хімічний зв'язок у комплексних з'єднаннях та їх будова У кристалічних комплексних з'єднаннях із зарядженими комплексами зв'язок між комплексом та зовнішньосферними іонами іонний, зв'язки між іншими частинками зовнішньої сфери – міжмолекулярні (у тому числі й водневі). У молекулярних комплексних сполуках зв'язок між комплексами міжмолекулярний. У більшості комплексних частинок між центральним атомом та лігандами зв'язки ковалентні. Усі вони або їх частина утворені за донорно-акцепторним механізмом (як наслідок – зі зміною формальних зарядів). У найменш міцних комплексах (наприклад, в аквакомплексах лужних та лужноземельних елементів, а також амонію) ліганди утримуються електростатичним тяжінням. Зв'язок у комплексних частках часто називають донорно-акцепторним або координаційним зв'язком. Розглянемо її утворення з прикладу аквакатиона заліза(II). Цей іон утворюється за реакцією: FeCl 2кр + 6H 2 O = 2 + 2Cl Електронна формула атома заліза. s 2 2s 2 2p 6 3s 2 3p 6 4s 2 3d 6 . Складемо схему валентних підрівнів цього атома: При утворенні двозарядного іона атом заліза втрачає два 4 s-електрона: Іон заліза акцептує шість електронних пар атомів кисню шести молекул води на вільні валентні орбіталі: Утворюється комплексний катіон, хімічну будову якого можна виразити однією з таких формул: Просторова будова цієї частки виражається однією з просторових формул: Форма координаційного поліедра – октаедр. Усі зв'язки Fe-O однакові. Передбачається sp 3 d 2-гібридизація АТ атома заліза. Магнітні властивості комплексу свідчить про наявність неспарених електронів. Якщо FeCl 2 розчиняти в розчині, що містить ціанід-іони, протікає реакція FeCl 2кр + 6CN = 4 + 2Cl. Той самий комплекс виходить і при додаванні до розчину FeCl 2 розчину ціаніду калію KCN: 2 + 6CN = 4 + 6H 2 O. Це говорить про те, що ціанідний комплекс міцніший за аквакомплекс. Крім того, магнітні властивості ціанідного комплексу вказують на відсутність неспарених електронів у атома заліза. Все це пов'язано з дещо іншою електронною будовою цього комплексу: Більш "сильні" ліганди CN утворюють міцніші зв'язки з атомом заліза, виграшу в енергії вистачає на те, щоб "порушити" правило Хунда і звільнити. d-орбіталі для неподілених пар лігандів Просторова будова ціанідного комплексу така сама, як і аквакомплексу, але тип гібридизації інший – d 2 sp 3 . "Сила" ліганду залежить насамперед від електронної щільності хмари неподіленої пари електронів, тобто вона збільшується зі зменшенням розміру атома, зі зменшенням головного квантового числа, залежить від типу гібридизації ЕО і від деяких інших факторів. Найважливіші ліганди можна побудувати до ряду за зростанням їх " сили " (своєрідний " ряд активності " лігандів), цей ряд називається спектрохімічним рядом лігандів: Для комплексів 3 та 3 схеми освіти виглядають наступним чином: Для комплексів з КЧ = 4 можливі дві структури: тетраедр (у разі sp 3-гібридизації), наприклад, 2 і плоский квадрат (у випадку dsp 2 -гібридизації), наприклад, 2 . 17.5. Хімічні властивості комплексних сполук Для комплексних сполук насамперед характерні самі властивості, як і звичайних сполук тих самих класів (солі, кислоти, основи). Якщо комплексне з'єднання кислота, це сильна кислота, якщо основа, те й основа сильне. Ці властивості комплексних сполук визначаються лише наявністю іонів H 3 O чи OH . Крім цього комплексні кислоти, основи та солі вступають у звичайні реакції обміну, наприклад: SO 4 + BaCl 2 = BaSO 4 + Cl 2 Остання з цих реакцій використовується як якісна реакція на іони Fe 3 . Нерозчинну речовину ультрамаринового кольору, що утворюється, називають "берлінською блакиттю" [систематична назва – гексаціаноферрат(II) заліза(III)-калію]. Крім цього реакцію може вступати і сама комплексна частка, причому, тим активніше, що вона менш стійка. Зазвичай це реакції заміщення лігандів, що протікають у розчині, наприклад: 2 + 4NH 3 = 2 + 4H 2 O, а також кислотно-основні реакції типу 2 + 2H 3 O = + 2H 2 O Утворюється в цих реакціях після виділення та висушування перетворюється на гідроксид цинку: Zn(OH) 2 + 2H 2 O Остання реакція – найпростіший приклад розкладання комплексної сполуки. У цьому випадку вона протікає за кімнатної температури. Інші комплексні з'єднання розкладаються при нагріванні, наприклад: SO 4 . H 2 O = CuSO 4 + 4NH 3 + H 2 O (вище 300 o С) Для оцінки можливості протікання реакції заміщення лігандів можна використовувати спектрохімічний ряд, керуючись тим, що сильніші ліганди витісняють із внутрішньої сфери менш сильні. 17.6. Ізомерія комплексних сполук Ізомерія комплексних сполук пов'язана До першої групи належить гідратна(у загальному випадку сольватна) та іонізаційнаізомерія, до другої – простороваі оптична. Гідратна ізомерія пов'язана з можливістю різного розподілу молекул води у зовнішній та внутрішній сферах комплексної сполуки, наприклад: (колір червоно-коричневий) та Br2 (колір блакитний). Іонізаційна ізомерія пов'язана з можливістю різного розподілу іонів у зовнішній та внутрішній сфері, наприклад: SO 4 (пурпурного кольору) та Br (червоного кольору). Перше з цих сполук утворює осад, реагуючи з розчином хлориду барію, а друге – з розчином нітрату срібла. Просторова (геометрична) ізомерія, інакше звана цис-транс ізомерією, характерна для квадратних та октаедричних комплексів (для тетраедричних неможлива). Приклад: цис-транс ізомерія квадратного комплексу Оптична (дзеркальна) ізомерія за своєю суттю не відрізняється від оптичної ізомерії в органічній хімії та характерна для тетраедричних та октаедричних комплексів (для квадратних неможлива).

Реакції координованих лігандів
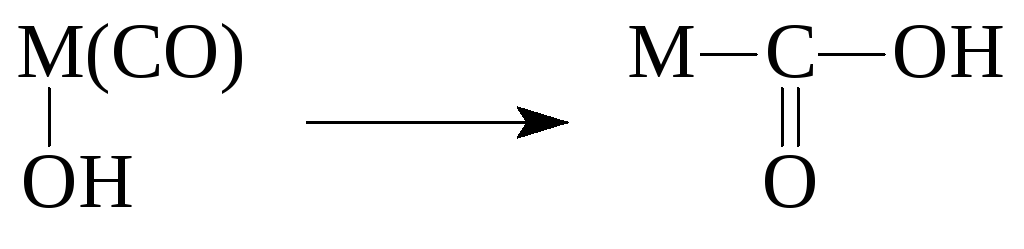








Реакції металоорганічних сполук





 Вторинна дисоціація -розпад внутрішньої сфери комплексу на складові її компоненти.
Вторинна дисоціація -розпад внутрішньої сфери комплексу на складові її компоненти.







BeSO 4 . 4H 2 O насправді SO 4;
Zn(BrO 3) 2 . 6H 2 O насправді (BrO 3) 2;
CuSO 4 . 5H 2 O насправді SO 4 . H2O.
AgBr + 2S 2 O 3 2 = 3 + Br
(При прояві фотоплівки та фотопаперу незасвітлена частина броміду срібла, що міститься у фотографічній емульсії, не відновлюється проявником. Для її видалення і використовують цю реакцію (процес носить назву "фіксування", тому що невіддалений бромід срібла надалі на світлі поступово розкладається, руйнуючи зображення)H 2 O - водний
Cl – хлоро
SO 4 2 – сульфато
OH – гідроксо
CO – карбоніл
Br – бромо
CO 3 2 – карбонато
H – гідридо
NH 3 – аммін
NO 2 – нітро
CN – ціано
NO - нітрозо
NO – нітрозил
O 2 – оксо
NCS – тіоціанато
H+I – гідро
3 – ді(тіосульфато)аргентат(I)-іон
3 – гексаціанохромат(III)-іон
– тетрагідроксодіакваалюмінат-іон
– тетранітродіаммінкобальтат(III)-іон
3 – пентаціаноакваферрат(II)-іон



I; Br; :
SCN, Cl, F, OH, H 2 O; :
NCS, NH 3; SO 3 S :
2 ; :
CN, CO


FeCl 3 + K 4 = Fe 4 3 + 3KCl
2 + 2OH = + 2H 2 O
4K 3 = 12KNO 2 + 4CoO + 4NO + 8NO 2 (понад 200 o С)
K 2 = K 2 ZnO 2 + 2H 2 O (вище 100 o С)
1) з можливим різним розташуванням лігандів та зовнішньосферних частинок,
2) з різною будовою найкомплекснішої частки.